

$ brew install sickle
$ brew install seqtk
$ java -jar ../../tools/Trimmomatic/trimmomatic-0.36.jar SE \
-phred33 -trimlog untreated.logfile \
raw_data/untreated.fq \
u.trimmomatic.fq \
SLIDINGWINDOW:5:20 LEADING:5 TRAILING:5 MINLEN:20
$ ~/software/com_extra/SOAPnuke/SOAPnuke1.5.6 filter -l 20 -G -Q 2 -1 untreated.fq -C u.soapnuke.fq.gz -o ./
-l 判定低质量碱基的阈值,它默认是5。我们这里为了保持和Trimmomatic的一致,同样设置为20;
-G 参数,如果设置了就表示质量值体系选择为phred33,默认是phred64。这是一个值得注意的地方,因为在SOAPnuke中是用sanger和illumina这两个词来分别代表phred33和phred64质量体系的。之所以会这样其实开发该软件的历史原因,在比较早期的时候,phred33和phred64这两个词用的还比较少。开发人员知道的是sanger测序的质量值是ASCII-33,而illumina的质量值要-64(早期版本),因此为了好记,就直接用了这两个词,代表和sanger的一样,或者和illumina的一样;
-Q 参数用于指明我们过滤后输出的read要选用哪种质量值体系输出,1代表phred64,2代表phred33,默认依然是phred33。SOAPnuke中的文档用的也依然是illumina和sanger作为不同质量值体系的标识;
-1 参数用于接输入的read1,相应的还有一个 -2 参数,用于接入read2,这是对于Pair-End测序而言的,我们这里只有一个fq,因此使用 -1 就可以了。
-C是输出文件的名字。SOAPnuke总是会输出gz压缩的文件,如果文件名中没有.gz,那么会被自动加上,如果没有指定这个-C参数,那么输出的文件会在原来输入文件的前面加上Clean_前缀;
-o 参数用于指定输出目录,默认是当前程序的运行目录;我们这里指定为当前目录;
SOAPnuke还有其它几个有用的参数,包括:-n代表一条read中可以允许多大比例的N;-d代表是否要去除PCR过程产生的重复序列,-q用于调整低质量碱基所占比例等等;此外,SOAPnuke还会在结果目录下同时输出其他几份文件,用于记录一下过滤前后的质量信息,由于本文中不会用到它们就不一一细说了。
$ sickle se -f raw_data/untreated.fq -t sanger -o u.sickle.fq
SE input file: raw_data/untreated.fq
Total FastQ records: 250000FastQ records kept: 249742FastQ records discarded: 258
$ seqtk trimfq raw_data/untreated.fq > u.trimfq.fqseqtk trimfq的功能比较单一,只能处理phred33,其他的都不行,参数很简洁。
【注意】 sickle和seqtk trimfq都不能用于adapter序列的切除,它们没有这个功能。
> source("https://bioconductor.org/biocLite.R")
> biocLite("qrqc")
library(qrqc)
# 指定fastq文件路径
fq_files = c(rawdata="raw_data/untreated.fq",
trimmomatic="u.trimmomatic.fq",
soapnuke="u.soapnuke.fq",
sickle="u.sickle.fq",
seqtk_trimfq="u.trimfq.fq")
# Load each fastq file in by using qrqc's readSeqFile
seq_read = lapply(fq_files, function(file) {
readSeqFile(file, hash = FALSE, kmer = FALSE)
})
quals = mapply(function(sfq, name) {
qs = getQual(sfq)
qs$trimmer = name
qs
}, seq_read, names(fq_files), SIMPLIFY = FALSE)
d = do.call(rbind, quals)
# 画图
library(ggplot2)
position = d$position
mean = d$mean
trimmer = d$trimmer
p1 = qplot(position, mean, color=trimmer, geom=c("line", "point"))
p1 = p1 + ylab("Mean quality (Phred33)") + theme_bw()
print(p1)
p2 = qualPlot(seq_read, quartile.color = NULL, mean.color = NULL) + theme_bw()
p2 = p2 + scale_y_continuous("Quality (Phred33)")
print(p2)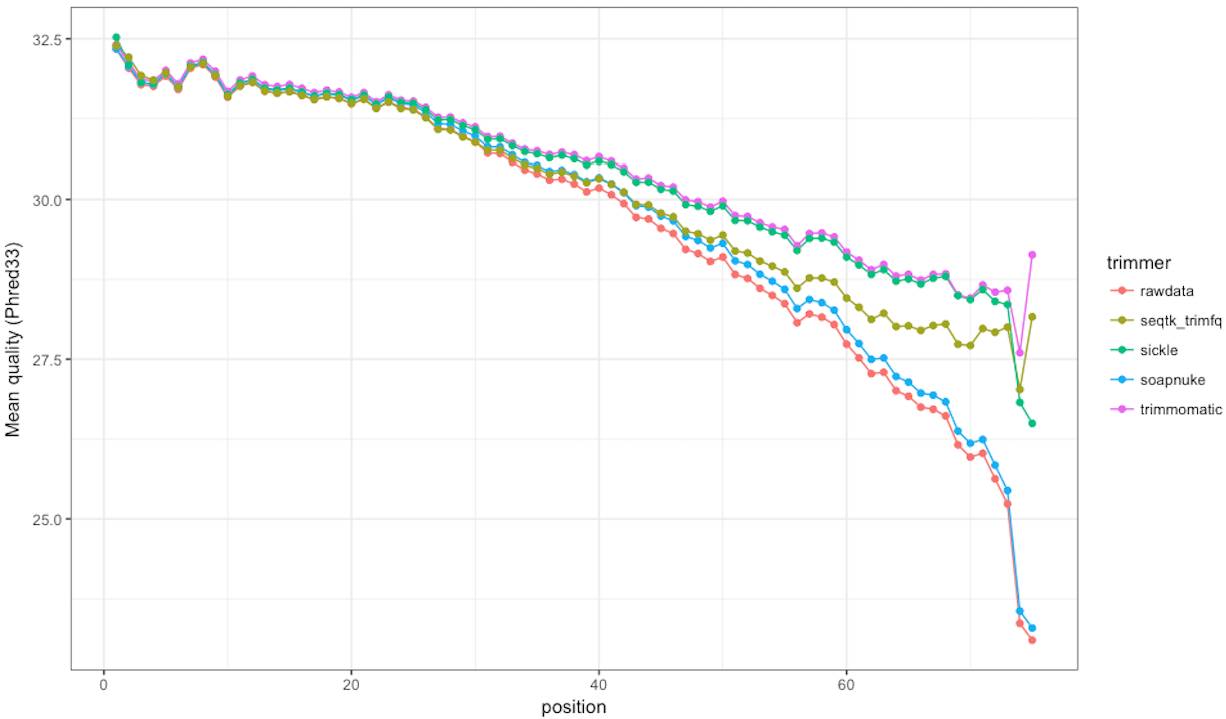
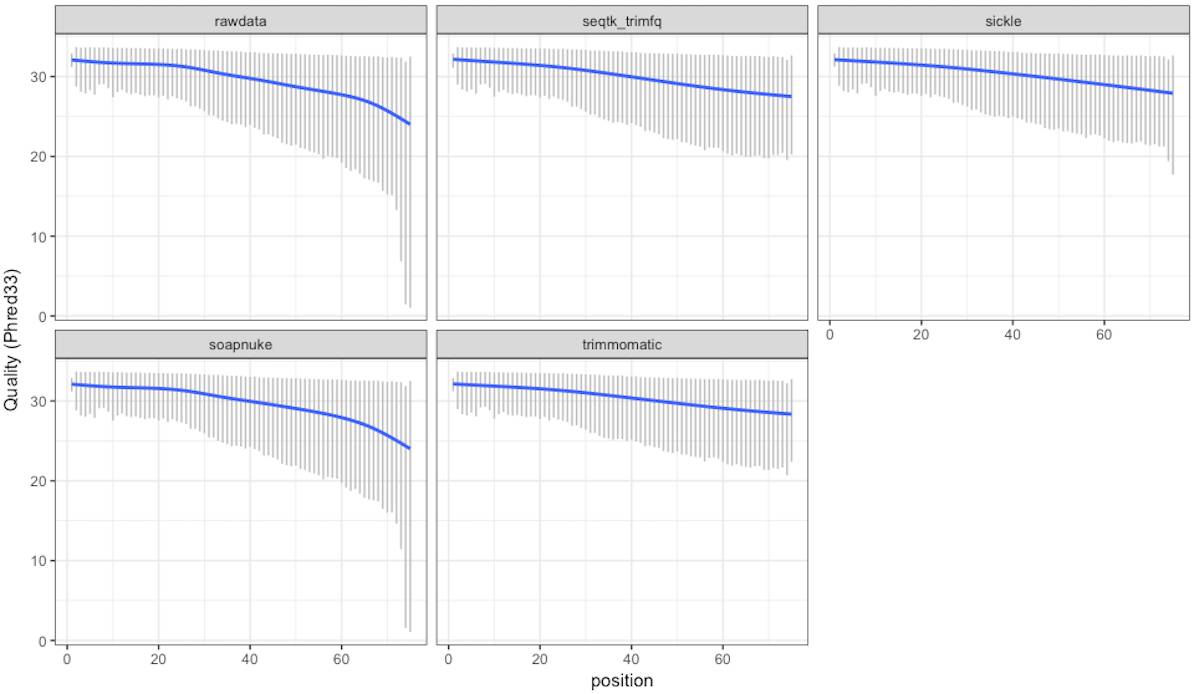
图2.
毫无疑问,从上面的比较结果中,我们可以明显发现,Trimmomatic最优,sickle次之,seqtk第三。当然,这其中结果最好的Trimmomatic丢弃掉的数据要比其他的略多(约1%~2%)。这三种里面比较特殊的是seqtk,它不丢弃任何read,只是将其截短,而且如果长度低于设定的最低read长度就不切除了,直接保留原数据。这也是为什么它相比于Trimmomatic和sickle而言质量要差的重要原因,可即便如此它们的结果其实相差并不多。反观SOAPnuke,不得不说SOAPnuke的过滤效果是最差的,几乎没什么效果。
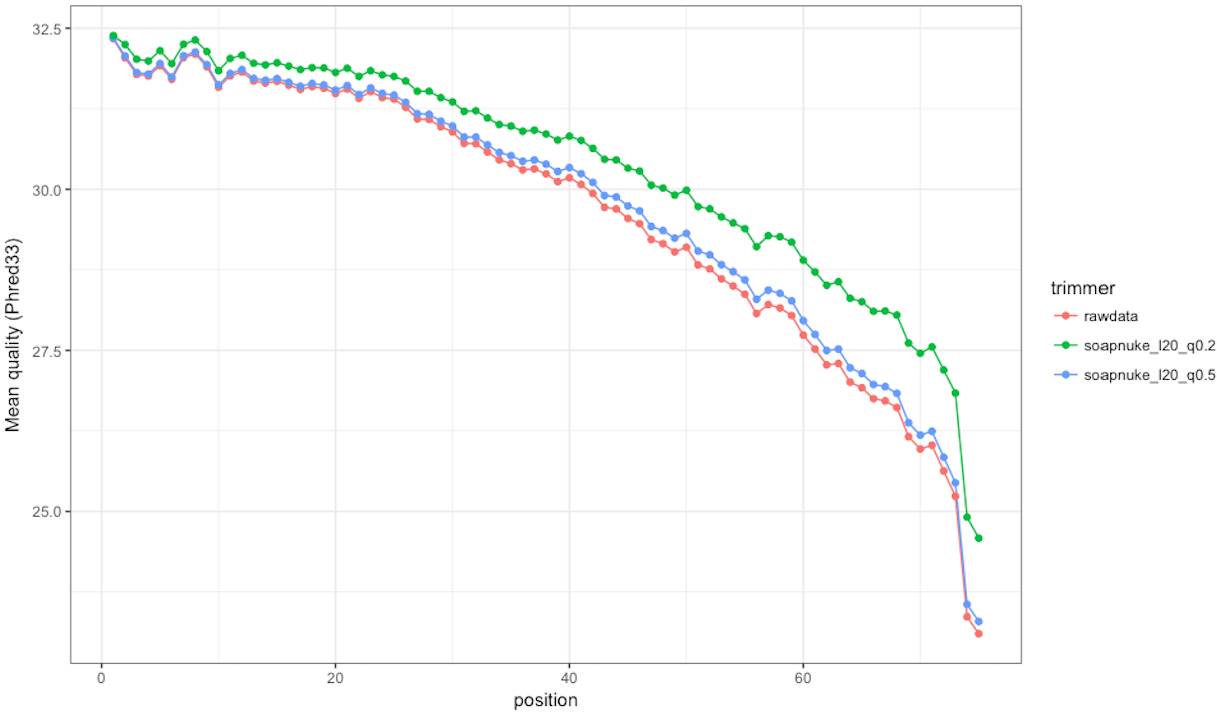
图3
这个图3中,我把SOAPnuke中低质量碱基占比的参数调得更加严格(20%)再和默认的50%相比,但发现改善并不特别明显。原因在于SOAPnuke并不切除read的末端低质量序列,而只是通过判断当read中低质量的碱基比例达到一定程度的时候整个read去除。但这种情况其实是不合理的,因为根据目前的测序原理,read的错误率会明显富集于末端区域,而前半部分的质量都会比较高,这种计算比例的方法并不能很好地反映这一现象。虽然我们可以通过设置更严格的参数,比如图3将低质量值碱基的占比从50%降为20%来改善,lowQual也可以设置得更高一些,来获得一些改善。但我想说的是,这种差别的根本原因源自于对末端低质量碱基序列的处理,SOAPnuke这种整体性统计的方法,并不能有针对性地处理这些碱基。在实际的操作要花更多的时间和精力对参数进行对比(影响最大的参数是-q低质量碱基占比),这不是一个高明的手段,远不如其他三个通过滑动窗口计算局部质量值的方式来得直接和灵敏。
综上,如果需要同时去除测序接头序列,那么建议使用Trimmomatic;如果只需过滤低质量碱基或者低质量read,可以选择Trimmomatic或sickle,有时sickle会更快一些,如果不愿意read被过滤掉而且质量值体系是phred33,那么可以选择seqtk。
------技术探索生命------
在这里与你分享专业而系统的NGS基因数据挖掘技术和方法

本文分享自微信公众号 - 碱基矿工(helixminer)。
如有侵权,请联系 support@oschina.cn 删除。
本文参与“OSC源创计划”,欢迎正在阅读的你也加入,一起分享。
来源:oschina
链接:https://my.oschina.net/u/4581491/blog/4372180